Precision Oncology in Practice: The Holy Grail of Prioritizing Oncogenic Alterations
Personalized medicine promises to match the right therapy to the right patient based on the unique genomic alterations driving their tumor. Yet despite remarkable progress, the majority of cancer patients still lack a clear path from sequencing results to targeted treatments. Here, we examine the interpretation bottleneck — and how evolving computational and clinical frameworks aim to close the gap between genomic data and real-world patient benefit.
What Are Mutations?
Cancer is not a single disease, but a dynamic ecosystem of evolving cells competing for survival. Tumor cells behave like opportunistic, parasite-like invaders: that is, adapting to selective pressures such as immune attack, nutrient scarcity, or even mechanical stress. Much like alien species introduced into new ecosystems, they exploit vulnerabilities and reshape their environment to thrive. Recent work even suggests that tumor cells can remember past stresses, enabling them to persist in the mechanically hostile environments of the bloodstream or distant metastatic sites.
Amid this complexity, genomic mutations remain a defining engine of tumorigenesis. But not all mutations are the same:
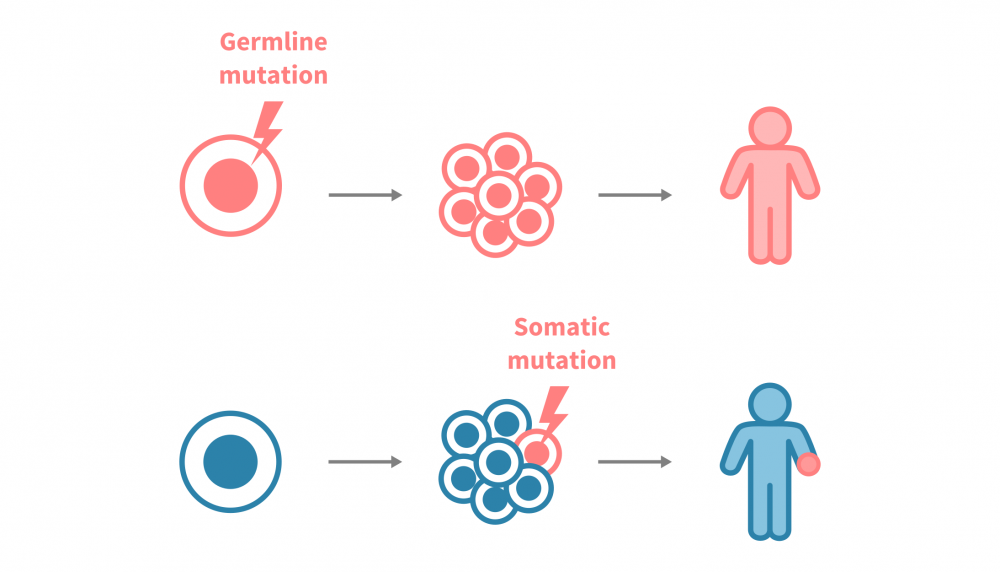
- Germline mutations are inherited from birth and passed on to offspring, while can be oncogenic: i.e., RB1 alterations predisposing to retinoblastoma.
- Somatic mutations arise in non-germ cells (e.g. somatic) during life. These acquired alterations (such as TP53 loss in many tumors) are not inherited, will not pass to offspring, but can profoundly shape cancer development.
The Promise and Reality of Precision Oncology
It is precisely these somatic alterations that lie at the heart of precision oncology. Over the last two decades, this approach has transformed cancer care, moving from one-size-fits-all chemotherapy toward targeted, biomarker-driven strategies. The premise is simple yet profound: by characterizing the genomic alterations that drive an individual’s tumor, clinicians can align therapy with biology, aiming to maximize efficacy while minimizing toxicity.
The impact has been already visible in various solid tumors. For example, EGFR mutations and MET exon 14 skipping, now guide therapy in non–small cell lung cancer (NSCLC). Likewise, HER2 amplification, and PIK3CA/ESR1 mutations inform treatment decisions in breast cancer. These models show that when actionable biomarkers are implemented, testing informs therapy and improves outcomes.
Yet despite these successes, the reality is sobering. Most cancer patients today still lack a clear, actionable alteration (personalized medicine benefits only about 25% of patients today). And even when detected, interpreting variants — not just calling them — is the critical bottleneck. Whole exome/genome sequencing (WES/WGS) routinely produces hundreds to thousands of somatic variants per patient, and the task is to distinguish drivers from passengers, and clinically actionable from biologically interesting but irrelevant.
Molecular tumor boards (MTBs) have emerged to bridge this gap, bringing together oncologists, molecular pathologists, bioinformaticians, and translational scientists to discuss complex cases. MTBs increasingly aim to incorporate multi-omic layers and longitudinal data. But they face structural limits: for example, they cannot scale indefinitely, and community centers often lack such expertise entirely. Cancer genome interpretation, therefore, is the process of analyzing the genetic changes in cancer cells to understand their role in tumor development and behavior, as well as to select the most effective therapy. It is the crucial step linking genomic data to patient care.
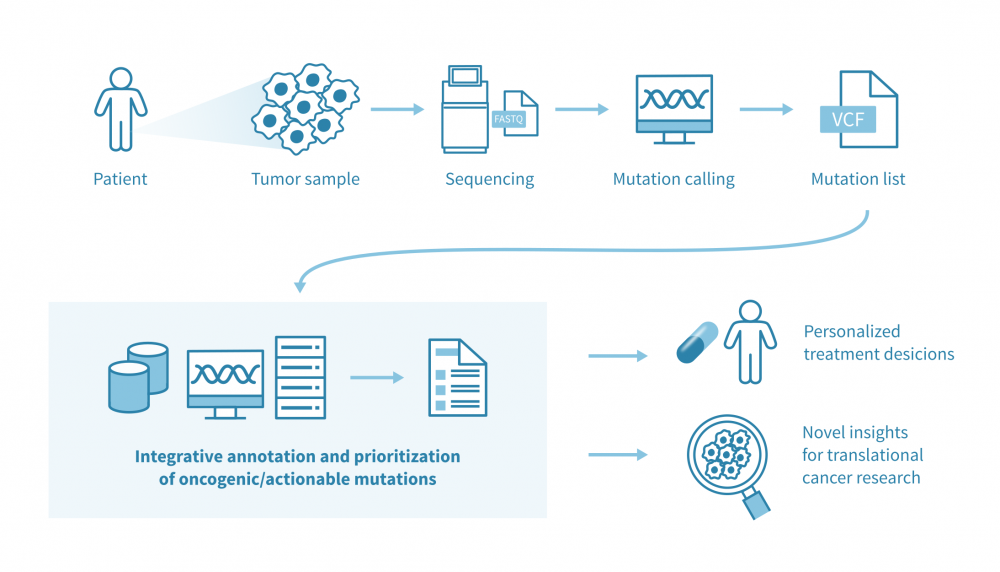
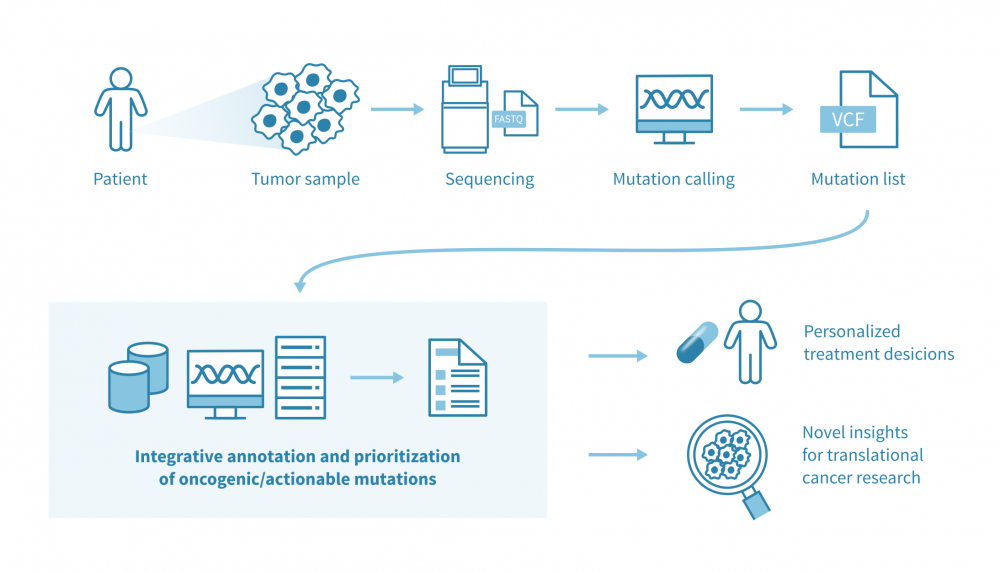
Main pipeline: From VCFs to ranked, interpretable alterations
Assumption: input VCFs already reflect somatic calls with standard artefact and germline filtering complete. From here, the task is to interpret systematically, reproducibly, and most importantly, ultimately to focus on patient resolution.
In this article we focus primarily on protein-coding variants, since most current annotation frameworks and clinical knowledge bases remain centered on coding alterations. Non-coding drivers are biologically important but remain harder to interpret in a standardized clinical context. Notable exceptions include recurrent promoter mutations (e.g., TERT) and new deep learning frameworks (e.g., Enformer, DeepSEA) that predict regulatory effects, but translation of such findings to practice is still challenging.
This integrative computational framework can be conceptualized in two major steps:
1. Multi-layered annotation as a foundation
Use a robust, modular, and standards-aware annotation platform (e.g., Ensembl VEP or OakVar) that can:
- Support standardized vocabularies (Sequence Ontology) and MANE transcripts: representative Matched Annotation from NCBI & EMBL-EBI (MANE) transcripts for one gene/variant, implementing a universal representation for clinical reporting, which increases the accuracy of variant interpretation.
- Provide genome-build liftover (hg19/hg38) and performant modular annotators.
- Aggregate heterogeneous evidence (population, pathogenicity, cancer knowledgebases, clinical links) in one place
- Population frequency filtering (e.g., gnomAD, ExAC) is crucial to (further) remove any common benign germline polymorphisms from somatic lists.
Why this matters: the quality and traceability of downstream prioritization depend directly on annotation depth and standardization.
2. Integrative filtering & prioritization: interpreting critical mutational circuits
Annotation alone is not interpretation. Variants should be filtered and prioritized by layering orthogonal evidence streams, rather than over-relying on any single predictor. Ultimately, robust prioritization frameworks integrate these layers into a transparent scoring or ranking scheme, based on the diversity of evidence considered. On this premise, a practical scheme that could be adopted accordingly, may group evidence into four complementary layers:
- Pathogenicity evidence
Functional impact of protein-coding variants (missense, truncating, splice-site, in-frame indels). Use ensemble or high-performing predictors like VEST4, PrimateAI, CHASMplus, MetaRNN, and splice effect predictors (i.e. SpliceAI), plus conservation/intolerance scores (e.g. PhyloP, PhastCons). - Cancer evidence
Signals of recurrence and tumor biology can be captured from both curated knowledgebases and computational discovery frameworks. Resources such as COSMIC, Cancer Hotspots, and IntOGen curate recurrent alterations and tumor-type–specific driver landscapes, integrating published evidence and large-scale cohort data. Complementing these, computational approaches like dNdScv identify statistically enriched drivers based on mutational spectra and selection signals. More recently, Oncodrive3D has extended this space by detecting spatial clustering of mutations in 3D protein structures, revealing functional driver hotspots across tumors. - Clinical support
Use ClinVar, OncoKB, CIViC, and other curated oncology knowledgebases, together with structured classification schemes (e.g., AMP/ASCO/CAP, ESMO ESCAT) and principles from large expert consortia such as the Variant Interpretation for Cancer Consortium (VICC). - Expression context
Check if the gene is expressed in the tumor using in-house data or public resources (e.g., TCGA/GTEx). Lack of expression can de-prioritize otherwise plausible hits; strong expression can lift borderline calls.
Also aim to assess the following contextual factors-if available-:
- Copy-number alterations (CNAs): multi-hits in a gene support oncogenicity.
- Variant allele frequency (VAF): clonal vs subclonal events.
- Tumor purity/ploidy estimates: to calibrate VAF and CNA interpretation.
Collectively, this multi-evidence framework prevents misinterpretation,e.g., over-weighting rare missense in tumor suppressors or undervaluing canonical activating oncogene mutations.
Challenges & Pitfalls
Even with careful annotation and filtering, conceptual hurdles remain:
- Passenger vs driver mutations: not always a clean binary. Some passengers contribute cumulatively to oncogenic pathways.
- Variants of unknown significance (VUS): common, especially in rare tumor types or under-characterized genes. Most will be non-actionable, but some may be critical.
- Tissue tropism: the same mutation can act as a driver in one tissue but not in another. Context matters.
- Rare cancers (i.e. MCC) and low mutational burden tumors (e.g. PDAC): small patient groups, complex biology and scattered infrastructure make it difficult to build robust training sets or apply population-level models.
Overall, these issues highlight why purely statistical driver-finding methods often fail at patient level: they overlook rare but actionable events and underperform with small cohorts.
Patient-level prioritization and reporting
A number of tools now attempt to bring all this together at the patient resolution. Interestingly, the most successful example is the Cancer Genome Interpreter (CGI): a notable state-of-the-art platform, and CGI-Clinics extends this through a community-driven effort to harmonize variant interpretation in clinical settings. Another interesting approach is the SVRACAS computational score: a prototype scheme created initially in collaboration with Johns Hopkins University and Oak Bioinformatics, LLC, for integrative somatic variant annotation and prioritization, tested both in public and in-house cancer datasets. SVRACAS integrates evidence from multiple (~15) biological and clinical resources into a unified ranking score, helping to contextualize the oncogenicity of alterations at the individual-patient level.
The Road Ahead: AI-Driven Clinical Decision Support
In the meantime, emerging directions are now expanding what variant interpretation can translate into:
- Proteomics-informed AI models are starting to enhance variant classification power: for example, integrating large-scale proteomics with large language models (LLMs) has been shown to improve rare missense variant classification and boost discovery of new gene–trait associations. This underscores the value of adding functional molecular layers to sharpen variant interpretation.
- AI-driven personalized oncology support systems are also gaining traction, aiming to assist MTBs and oncologists in making evidence-based decisions at scale. These tools promise to prioritize transparency and explainability to overcome the early “black box” bottleneck (see example).
- Finally, liquid biopsy (ctDNA) profiling is becoming a critical complement to tissue genomics: enabling real-time monitoring of tumor evolution, detection of emerging resistance, evaluation of minimal residual disease, and early identification of recurrence well before it becomes clinically or radiographically apparent. Together, these innovations hint at a future where precision oncology is not just reactive but proactive, anticipating tumor behavior and tailoring care dynamically over time.
The author is a co-developer of the above-mentioned SVRACAS — Learn more about this scoring scheme in the published article.
Contact us
Leave your email address here with a brief description of your needs, and we will contact you to get things moving forward!
