RNA sequencing data analysis

RNA sequencing data analysis brings to light the intricate mechanisms of gene regulation.
Transcriptome-wide analyses of gene expression are extremely popular among researchers studying gene regulation in biological systems ranging from single cells to tissues and complex microbiomes. RNA-seq data allows for a wide range of analyses to address countless research questions across the fields of biology and biomedicine.
Below we present some of the most common analyses we perform on RNA-seq data. The explorative, differential expression and pathway analyses largely apply to other high-throughput expression data as well, such as expression microarray or proteomic data.
We hope that the examples below inspire you to appreciate just how rich the world of RNA-sequencing is. If you are planning an RNA-seq experiment and wish to learn how we can help you to get the most out of your data, leave us a message and we will book you a short call with our expert.
Leave us a short description of your bioinformatics needs and we will be in touch very soon!
Exploratory gene expression analysis
Every RNA-seq expression study incorporates an exploratory analysis. After the raw sequencing reads of an RNA-seq experiment have been quality controlled and gene counts derived, the data set is visualized using Principal Component Analysis (PCA) and expression heatmaps to unveil its general patterns. These visualizations help us answer questions such as:
- Do the biological replicates resemble each other with regards to their expression profiles?
- Do distinct sample groups (e.g., different tissues, treatments or time points) form separate clusters?
- Are there outlier samples?

Differential expression analysis
Differential expression analysis is a statistical comparison of two sample groups. It results in differential expression statistics for each detected transcript, such as the fold change and statistical significance. These statistics are typically visualized using a volcano plot. The genes which are found to be up- or down-regulated can be further visualized as heatmaps or boxplots, for instance.
As a statistical analysis, this phase of an expression study benefits from the statistical power brought by biological replicates. Three biological replicates per condition is a common “rule-of-thumb” minimum, but it only allows for reliable detection of genes with relatively large expression differences. With a careful experimental design and sufficient sample size, subtler differences can be detected and confounding factors controlled for.

Pathway analysis
Pathway analysis puts genes from a differential expression analysis into broader biological context. Simple pathway analyses compare the up- and down-regulated genes statistically to predetermined gene lists. These lists are annotated to biologically meaningful terms, such as a biological process, signaling pathway or a specific disease.
Such analyses may rely either on over-representation analysis or gene set enrichment analysis, which both result in a list of enriched gene sets with relevant statistics and annotations.
More mechanistic pathway analyses rely on experimentally validated interactions between genes. They enable identifying not just which pathways are represented by the differentially expressed genes, but also shed light on whether the pathways are activated or inhibited, and by which genes.
For the more avanced pathway analyses, we use Ingenuity Pathway Analysis (IPA, QIAGEN). IPA enables a wide range of in-depth analyses into known and novel gene regulatory networks.

Transcriptome assembly
For non-model organisms, and those with very dynamic genomes, i.e. microbes, we typically start RNA sequencing data analysis with assembling a transcriptome de novo and annotating it using homologues of related species and computational gene predictions.
A new reference transcriptome is an invaluable resource for your further research, and that of the entire research community. Once a high-quality reference transcriptome has been established, the door opens to most downstream analyses which are routinely used with model organisms.
![]()
Single-cell expression analysis
Single-cell RNA-sequencing (scRNA-seq) experiments allow for cataloguing cell types and uncovering differentiation trajectories at a scale and resolution unmatched by bulk RNA sequencing.
Used particularly to study the composition and development of complex tissues, scRNA-seq data sets typically comprise thousands of individual cells. Most approaches used to analyze bulk RNA-seq data can be tailored for single-cell RNA-seq data as well.

MicroRNA data analysis
Small RNA-sequencing enables studying various species of short RNAs, and microRNAs in particular. MicroRNA-seq analysis is largely similar to that of mRNAs, but pathway and regulatory analyses make use of predicted and/or previously validated microRNA target genes.
Sequencing both mRNA and small RNA from the matched samples enables estimating the regulatory relationship between microRNAs and their targets. To identify genes subject to microRNA-mediated regulation in a given condition, argonaute CLIP-sequencing (and related protocols) can be employed.

Alternative splicing analysis
In addition to studying expression on the level of genes, RNA-sequencing allows for a more detailed view: splice-variant level expression. Reliable identification of alternative splicing events benefits from deeper sequencing than the typical gene-level expression analysis.
Depending on the quantity and quality of the data, alternative splicing analyses may focus on quantifying expression levels of known, previously annotated splice isoforms, or on detecting novel splicing events as well.

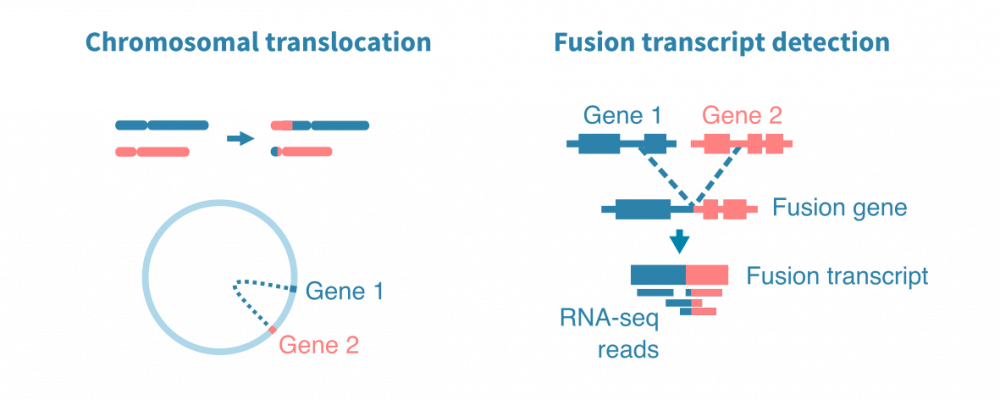
Fusion gene detection
In cancer, certain structural variants are known to cause fusion genes. Two separate genes fused together in the DNA may lead to a fusion transcript. The fusion transcript, in turn, may lead to a fusion protein with a novel, potentially cancer-driving combination of regulation and function.
Fusion genes can be detected from RNA-sequencing data with tools that identify and analyze discordantly mapping RNA-seq reads or read pairs.
Integrating RNA-seq and epigenomic data
Performing RNA-seq and epigenomic sequencing (such as ChIP or ATAC-seq) on the same samples enables integrative analyses to study gene regulatory programs genome-wide.
Regulatory connections can be identified between enhancers and their target genes, as well as transcription factors and their targets, building on evidence from both gene expression and the epigenomic status of regulatory elements.

Meet some of our transcriptomics experts
I am a senior molecular biologist and bioinformatician with more than 8 years of experience with molecular techniques and NGS data analysis. My research background straddles many fields including neurobiology, aging, immunology, virology, microbiology, and aquaculture.
I have expertise in a broad suite of bioinformatics tools for diverse analyses including variant analysis (SNP arrays, WGS, RNaseq), differential expression and co-expression (mRNAseq, 3’/TAGseq), and de novo genome and transcriptome assembly and annotation. I have worked with diverse organisms from across the tree of life, including viruses, single celled algae, corals, mollusks, fish, and human.
My educational background in comparative and molecular biology, experience with molecular techniques, and expertise in bioinformatic analysis allow me to effectively integrate and collaborate with scientists and non-scientists from diverse backgrounds.

I am a senior bioinformatician with extensive experience in analysing data types including mRNA-seq, smallRNA-seq, DNA-seq, ChIP-seq, proteomics and methylation data.
During the years working on customer projects I have gained experience in e.g. gene and protein expression analysis, germline and somatic variant analysis, genome-wide association studies and polygenic risk score analysis, population genomics, epigenetic analysis, using public ‘omics datasets such as TCGA and applying machine learning methods for patient prognosis.
I am also very experienced in leveraging the various pathway and gene-regulatory analyses of the Ingenuity Pathway Analysis (IPA) software. In my data analysis role, I also benefit from my 10+ years of experience as a wet-lab molecular biologist as well as my experience in biocuration and database content creation.

As a scientist, I specialize in cellular differentiation and RNA biology. I have been studying the interplay between transcriptional regulators and non-coding RNAs in a multitude of biomedical contexts, including mesenchymal stem cell differentiation, endothelial cell differentiation, atherosclerosis and leukemia.
From the methodological perspective, I have been trained as a comprehensive systems biologist and generated NGS datasets myself in my research projects. I am experienced in analyzing sequencing data from RNA (mRNA-seq, short RNA-seq, scRNA-seq, GRO-seq, TT-seq) and DNA libraries (ChIP-seq, ATAC-seq, CITE-seq) and in integrating different data modalities to gain ever deeper insight into complex systems.

Learn more
The examples above showcased some of the more typical computational analyses with RNA-seq data. The opportunitites with RNA-seq, however, are nearly endless, as is our team's experience!
Learn more about RNA-seq data analysis
- Single-cell RNA-seq data analysis
- Spatial transcriptomic data analysis
- On interpreting RNA-seq data analysis results
- On predicting cancer survival from tumor RNA-seq data
References and customer cases
- Decoding STAT1 Isoform-Specific Control of Macrophage Responses Through Multiomics Analysis (University of Veterinary Medicine Vienna)
- Integrating scRNA-Seq and CUT&Tag data to investigate genomic instability in aging models (University of Cologne)
- Characterizing a cancer immunotherapy target with genomic data (CDR-Life)
- Studying neural circuit development with single-cell RNA-sequencing (Cornell University)
- Investigating a Parkinson's disease treatment's mechanism of action using bulk and spatial transcriptomics (Herantis Pharma)
- Deciphering progression mechanisms in chronic kidney disease with single-nucleus and spatial transcriptomics (University of Geneva)
- Investigating vector-borne diseases and pathogen transmission biology with transcriptomics (University of Florida)Building a neuronal atlas with single-cell technologies (VIB)
- Virtual bioinformatics core service for immunology research (University of Western Australia)Discovering cell markers and characterizing microglial response to viral infection (University of Fribourg)
- Tailored bioinformatics support for a clinical trial (Amazentis)
- Whole-genome and RNA sequencing analyses of hereditary cancer (University of Turku)Single-cell expression analysis (Becton Dickinson)
- Transcriptomic cell-therapy biomarker discovery (Kuopio Center for Gene and Cell Therapy)
- Livestock feed additive mode of action study (Alimetrics)
- Transcriptome-based cancer target identification (Barron Biomedical)
- Multi-omic biomarker analysis (FastEV)
- Multi-omic mechanism of action study (MinaTx)
- Investigating the role of glypicans in cancer
Selected publications from our customers
- Onyenobi, E. et al. (2026). Identification of miRNA biomarkers and development of predictive signatures for early detection and prognosis in cervical precancer and cancer. Human genomics, 20(1), 60. https://doi.org/10.1186/s40246-026-00924-z
- Zhong, M. et al. (2026). Novel circRNA signatures in cervical cancer: implications for early detection and screening. Biomarkers in medicine, 20(2), 119–128. https://doi.org/10.1080/17520363.2026.2631965
- Akagha, M. J. et al. (2026). IFNγ shapes macrophage inflammatory responses by STAT1 isoform-specific epigenetic and transcriptional mechanisms. BMC genomics, 10.1186/s12864-026-12601-5. Advance online publication. https://doi.org/10.1186/s12864-026-12601-5
- Gehrs, S. et al. (2025). The spatial zonation of the murine placental vasculature is specified by epigenetic mechanisms. Developmental cell, S1534-5807(24)00776-7. Advance online publication. https://doi.org/10.1016/j.devcel.2024.12.037
- Liu, S. et al. (2025). Urolithin A provides cardioprotection and mitochondrial quality enhancement preclinically and improves human cardiovascular health biomarkers. iScience, 28(2), 111814. https://doi.org/10.1016/j.isci.2025.111814
- Zitta, K. et al. (2025). Transcriptomic characterization of GMP-compliant regulatory macrophages (TRI-001) under inflammatory and hypoxic conditions: a comparative analysis across macrophage subtypes. Journal of translational medicine, 23(1), 551. https://doi.org/10.1186/s12967-025-06548-6
Velecela, V. et al. (2025). WNT inhibition primes the transcriptional landscape of mesoderm to initiate a phased ventricular cardiomyocyte specification programme. bioRxiv 2025.11.11.687613; doi: https://doi.org/10.1101/2025.11.11.687613
Barreiro, K. et al. (2025). Selected miRNAs in Urinary Extracellular Vesicles Show Promise for Early and Specific Diagnostics of Diabetic Kidney Disease. Journal of extracellular biology, 4(10), e70089. https://doi.org/10.1002/jex2.70089
Soboleva, T. et al. (2025). Histone Variant H2A.B.3 Orchestrates a New Nuclear Pathway for Histone mRNA Decay. Research Square preprint https://doi.org/10.21203/rs.3.rs-8304477/v1
Mallikarjuna, P. et al. (2025). Liquid biomarkers associate with TGF-β Type I receptor and hypoxia in kidney cancer. Signal transduction and targeted therapy, 10(1), 309. https://doi.org/10.1038/s41392-025-02404-7
Roland, V. et al. (2025). Functional architecture of cardiac TF regulatory landscapes in control of mammalian heart development. bioRxiv 2025.12.19.695499; doi: https://doi.org/10.64898/2025.12.19.695499
- Chang, Y. T. et al. (2024). MHC-I upregulation safeguards neoplastic T cells in the skin against NK cell-mediated eradication in mycosis fungoides. Nature communications, 15(1), 752. https://doi.org/10.1038/s41467-024-45083-8
- Peeters, J. G. C. et al. (2024). Hyperactivating EZH2 to augment H3K27me3 levels in regulatory T cells enhances immune suppression by driving early effector differentiation. Cell reports, 43(9), 114724. Advance online publication. https://doi.org/10.1016/j.celrep.2024.114724
- Ambite, I. et al. (2024). Molecular analysis of acute pyelonephritis—excessive innate and attenuated adaptive immunity. Life Science Alliance, 8(3), e202402926. https://doi.org/10.26508/lsa.202402926
- Schmidt-Christensen, A. et al. (2024). Structure-function analysis of time-resolved immunological phases in metabolic dysfunction-associated fatty liver disease (MASH) comparing the NIF mouse model to human MASH. Scientific reports, 14(1), 23014. https://doi.org/10.1038/s41598-024-73150-z
- Häyrinen, M. et al. (2024). Tumor RNA sequencing identifies a group of Mycosis Fungoides patients with failure of skin-directed therapies. The Journal of investigative dermatology, S0022-202X(24)02084-0. Advance online publication. https://doi.org/10.1016/j.jid.2024.06.1292
- Nissinen, L. et al. (2024). Clustering of RNA co-expression network identifies novel long non-coding RNA biomarkers in squamous cell carcinoma. Scientific reports, 14(1), 16864. https://doi.org/10.1038/s41598-024-67808-x
- Chérouvrier Hansson, V. et al. (2024). Dichotomous Effects of Glypican-4 on Cancer Progression and Its Crosstalk with Oncogenes. International journal of molecular sciences, 25(7), 3945. https://doi.org/10.3390/ijms25073945
- Fisher, J. et al. (2023). Cortical somatostatin long-range projection neurons and interneurons exhibit divergent developmental trajectories. Neuron, S0896-6273(23)00887-5. Advance online publication. https://doi.org/10.1016/j.neuron.2023.11.013
- D’Amico, D. et al. (2023). Topical application of Urolithin A slows intrinsic skin aging and protects from UVB-mediated photodamage: Findings from Randomized Clinical Trials. medRxiv 2023.06.16.23291378. https://doi.org/10.1101/2023.06.16.23291378
- Cheng, F. et al. (2023). Attenuation of cancer proliferation by suppression of glypican-1 and its pleiotropic effects in neoplastic behavior. Oncotarget, 14, 219–235. https://doi.org/10.18632/oncotarget.28388
- Häyrinen, M. J. et al. (2023). The Transcription Factor Twist1 Has a Significant Role in Mycosis Fungoides (MF) Cell Biology: An RNA Sequencing Study of 40 MF Cases. Cancers, 15(5), 1527. https://doi.org/10.3390/cancers15051527
- Singh, A. et al. (2022). Urolithin A improves muscle strength, exercise performance, and biomarkers of mitochondrial health in a randomized trial in middle-aged adults. Cell reports. Medicine, 3(5), 100633. https://doi.org/10.1016/j.xcrm.2022.100633
- Pihlström, S. et al. (2022). A multi-omics study to characterize the transdifferentiation of human dermal fibroblasts to osteoblast-like cells. Frontiers in molecular biosciences, 9, 1032026. https://doi.org/10.3389/fmolb.2022.1032026
- Tusup, M. et al. (2022). Epitranscriptomics modifier pentostatin indirectly triggers Toll-like receptor 3 and can enhance immune infiltration in tumors. Molecular therapy : the journal of the American Society of Gene Therapy, 30(3), 1163–1170. https://doi.org/10.1016/j.ymthe.2021.09.022
- Cramer, M. et al. (2022). Transcriptomic Regulation of Macrophages by Matrix-Bound Nanovesicle-Associated Interleukin-33. Tissue engineering. Part A, 28(19-20), 867–878. https://doi.org/10.1089/ten.TEA.2022.0006
- Pommergaard, H. C. et al. (2022). Aldehyde dehydrogenase expression may be a prognostic biomarker and associated with liver cirrhosis in patients resected for hepatocellular carcinoma. Surgical oncology, 40, 101677. https://doi.org/10.1016/j.suronc.2021.101677
- Song, J. et al. (2022). The ubiquitin-ligase TRAF6 and TGFβ type I receptor form a complex with Aurora kinase B contributing to mitotic progression and cytokinesis in cancer cells. EBioMedicine, 82, 104155. https://doi.org/10.1016/j.ebiom.2022.104155
- Martins, R. R. et al. (2022). Trancriptomic signatures of telomerase-dependent and -independent ageing, in the zebrafish gut and brain. bioRxiv 2022.05.24.493215; doi: 101677. https://doi.org/10.1101/2022.05.24.493215
- Kundu, S. et al. (2021). Common and mutation specific phenotypes of KRAS and BRAF mutations in colorectal cancer cells revealed by integrative -omics analysis. Journal of experimental & clinical cancer research : CR, 40(1), 225. https://doi.org/10.1186/s13046-021-02025-2
- Pommergaard, H. C. et al. (2021). Peroxisome proliferator-activated receptor activity correlates with poor survival in patients resected for hepatocellular carcinoma. Journal of hepato-biliary-pancreatic sciences, 28(4), 327–335. https://doi.org/10.1002/jhbp.745
- Lehto, T. K. et al. (2021). Transcript analysis of commercial prostate cancer risk stratification panels in hard-to-predict grade group 2-4 prostate cancers. The Prostate, 81(7), 368–376. https://doi.org/10.1002/pros.24108
- Hussey, G. S. et al. (2020). Lipidomics and RNA sequencing reveal a novel subpopulation of nanovesicle within extracellular matrix biomaterials. Science advances, 6(12), eaay4361. https://doi.org/10.1126/sciadv.aay4361
- Oksanen, M. et al. (2020). NF-E2-related factor 2 activation boosts antioxidant defenses and ameliorates inflammatory and amyloid properties in human Presenilin-1 mutated Alzheimer's disease astrocytes. Glia, 68(3), 589–599. https://doi.org/10.1002/glia.23741
- Lemke, P. et al. (2020). Transcriptome Analysis of Solanum Tuberosum Genotype RH89-039-16 in Response to Chitosan. Frontiers in plant science, 11, 1193. https://doi.org/10.3389/fpls.2020.01193
- Tiihonen, J. et al. (2020). Neurobiological roots of psychopathy. Molecular psychiatry, 25(12), 3432–3441. https://doi.org/10.1038/s41380-019-0488-z
- Gurvich, O. L. et al. (2020). Transcriptomics uncovers substantial variability associated with alterations in manufacturing processes of macrophage cell therapy products. Scientific reports, 10(1), 14049. https://doi.org/10.1038/s41598-020-70967-2
- Gabriel, M. et al. (2020). A relational database to identify differentially expressed genes in the endometrium and endometriosis lesions. Scientific data, 7(1), 284. https://doi.org/10.1038/s41597-020-00623-x
- Tiihonen, J. et al. (2019). Sex-specific transcriptional and proteomic signatures in schizophrenia. Nature communications, 10(1), 3933. https://doi.org/10.1038/s41467-019-11797-3
- Tarkkonen, K et al. (2017). Comparative analysis of osteoblast gene expression profiles and Runx2 genomic occupancy of mouse and human osteoblasts in vitro. Gene, 626, 119–131. https://doi.org/10.1016/j.gene.2017.05.028
- Sugano, Y. et al. (2017). Comparative transcriptomic analysis identifies evolutionarily conserved gene products in the vertebrate renal distal convoluted tubule. Pflugers Archiv : European journal of physiology, 469(7-8), 859–867. https://doi.org/10.1007/s00424-017-2009-8
Selected publications from our team
- Psatha, N. et al. (2025). Large-scale discovery of potent, compact and erythroid specific enhancers for gene therapy vectors. Nature communications, 16(1), 4325. https://doi.org/10.1038/s41467-025-59235-x
- Brunner, E. et al. (2025). Unraveling the YAP1-TGFβ1 axis: a key driver of androgen receptor loss in prostate cancer-associated fibroblasts. bioRxiv 2025.02.25.640167; doi: https://doi.org/10.1101/2025.02.25.640167
- Punzon-Jimenez, P. et al. (2024). Effect of aging on the human myometrium at single-cell resolution. Nature communications, 15(1), 945. https://doi.org/10.1038/s41467-024-45143-z
- Fotakis, G. et al. (2024). Conventional therapy induces tumor immunoediting and modulates the immune contexture in colorectal cancer. bioRxiv 2024.08.21.608938; doi: https://doi.org/10.1101/2024.08.21.608938
- Kron, N. S. et al. (2024). Expression dynamics of the aplysia abyssovirus. Virology, 589, 109890. https://doi.org/10.1016/j.virol.2023.109890
- Lin, J. et al. (2023). Distinct transcriptomic profiles in children prior to the appearance of type 1 diabetes-linked islet autoantibodies and following enterovirus infection. Nature communications, 14(1), 7630. https://doi.org/10.1038/s41467-023-42763-9
- Caronni, N. et al. (2023). IL-1β+ macrophages fuel pathogenic inflammation in pancreatic cancer. Nature, 623(7986), 415–422. https://doi.org/10.1038/s41586-023-06685-2
- Beumers, L. et al. (2023). Clonal heterogeneity in ER+ breast cancer reveals the proteasome and PKC as potential therapeutic targets. NPJ breast cancer, 9(1), 97. https://doi.org/10.1038/s41523-023-00604-4
- Simigdala, N. et al. (2023). Loss of Kmt2c in vivo leads to EMT, mitochondrial dysfunction and improved response to lapatinib in breast cancer. Cellular and molecular life sciences : CMLS, 80(4), 100. https://doi.org/10.1007/s00018-023-04734-7
- Korvenlaita, N. et al. (2023). Dynamic release of neuronal extracellular vesicles containing miR-21a-5p is induced by hypoxia. Journal of extracellular vesicles, 12(1), e12297. https://doi.org/10.1002/jev2.12297https://doi.org/10.1002/jev2.12297
- Saralahti, A. K. et al. (2023). Characterization of the innate immune response to Streptococcus pneumoniae infection in zebrafish. PLoS genetics, 19(1), e1010586. Advance online publication. https://doi.org/10.1371/journal.pgen.1010586
- Aakula, A. et al. (2023). RAS and PP2A activities converge on epigenetic gene regulation. Life science alliance, 6(5), e202301928. https://doi.org/10.26508/lsa.202301928
- Kontogianni, G. et al. (2023). A Comprehensive Analysis of Cutaneous Melanoma Patients in Greece Based on Multi-Omic Data. Cancers, 15(3), 815. https://doi.org/10.3390/cancers15030815
- Pham, T. et al. (2022). Modeling human extraembryonic mesoderm cells using naive pluripotent stem cells. Cell stem cell, 29(9), 1346–1365.e10. https://doi.org/10.1016/j.stem.2022.08.001
- Zijlmans, D. W. et al. (2022). Integrated multi-omics reveal polycomb repressive complex 2 restricts human trophoblast induction. Nature cell biology, 24(6), 858–871. https://doi.org/10.1038/s41556-022-00932-w
- Smith, C. et al. (2022). A comparative transcriptomic analysis of glucagon-like peptide-1 receptor- and glucose-dependent insulinotropic polypeptide-expressing cells in the hypothalamus. Appetite, 174, 106022. https://doi.org/10.1016/j.appet.2022.106022
Heidegger, I. et al. (2022). Comprehensive characterization of the prostate tumor microenvironment identifies CXCR4/CXCL12 crosstalk as a novel antiangiogenic therapeutic target in prostate cancer. Molecular cancer, 21(1), 132. https://doi.org/10.1186/s12943-022-01597-7
- Cao, S. et al. (2022). Estimation of tumor cell total mRNA expression in 15 cancer types predicts disease progression. Nature biotechnology, 10.1038/s41587-022-01342-x. Advance online publication. https://doi.org/10.1038/s41587-022-01342-x
- Montaldo, E. et al. (2022). Cellular and transcriptional dynamics of human neutrophils at steady state and upon stress. Nature immunology, 23(10), 1470–1483. https://doi.org/10.1038/s41590-022-01311-1
- Gao, Y. et al. (2022). Inactivation of AMPK Leads to Attenuation of Antigen Presentation and Immune Evasion in Lung Adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research, 28(1), 227–237. https://doi.org/10.1158/1078-0432.CCR-21-2049
- Rodriguez-Martinez, A. et al. (2022). Novel ZNF414 activity characterized by integrative analysis of ChIP-exo, ATAC-seq and RNA-seq data. Biochimica et biophysica acta. Gene regulatory mechanisms, 1865(3), 194811. Advance online publication. https://doi.org/10.1016/j.bbagrm.2022.194811
Rieder, D. et al. (2022). nextNEOpi: a comprehensive pipeline for computational neoantigen prediction. Bioinformatics (Oxford, England), 38(4), 1131–1132. https://doi.org/10.1093/bioinformatics/btab759
Roos, K. et al. (2022). Single-cell RNA-seq analysis and cell-cluster deconvolution of the human preovulatory follicular fluid cells provide insights into the pathophysiology of ovarian hyporesponse. Frontiers in endocrinology, 13, 945347. https://doi.org/10.3389/fendo.2022.945347
Pellegrinelli, V. et al. (2022). Dysregulation of macrophage PEPD in obesity determines adipose tissue fibro-inflammation and insulin resistance. Nature metabolism, 4(4), 476–494. https://doi.org/10.1038/s42255-022-00561-5
Kukkonen, K. et al. (2022). Nonmalignant AR-positive prostate epithelial cells and cancer cells respond differently to androgen. Endocrine-related cancer, 29(12), 717–733. https://doi.org/10.1530/ERC-22-0108
Georgolopoulos, G. et al. (2021). Discrete regulatory modules instruct hematopoietic lineage commitment and differentiation. Nature communications, 12(1), 6790. https://doi.org/10.1038/s41467-021-27159-x
- Taavitsainen, S. et al. (2021). Single-cell ATAC and RNA sequencing reveal pre-existing and persistent cells associated with prostate cancer relapse. Nature communications, 12(1), 5307. https://doi.org/10.1038/s41467-021-25624-1
- Cilenti, F. et al. (2021). A PGE2-MEF2A axis enables context-dependent control of inflammatory gene expression. Immunity, 54(8), 1665–1682.e14. https://doi.org/10.1016/j.immuni.2021.05.016
- Linna-Kuosmanen, S. et al. (2021). NRF2 is a key regulator of endothelial microRNA expression under proatherogenic stimuli. Cardiovascular research, 117(5), 1339–1357. https://doi.org/10.1093/cvr/cvaa219
- Moreau, P. R. et al. (2021). Profiling of Primary and Mature miRNA Expression in Atherosclerosis-Associated Cell Types. Arteriosclerosis, thrombosis, and vascular biology, 41(7), 2149–2167. https://doi.org/10.1161/ATVBAHA.121.315579
- Carobbio, S. et al. (2021). Unraveling the Developmental Roadmap toward Human Brown Adipose Tissue. Stem cell reports, 16(3), 641–655. https://doi.org/10.1016/j.stemcr.2021.01.013
- Filppu, P. et al. (2021). CD109-GP130 interaction drives glioblastoma stem cell plasticity and chemoresistance through STAT3 activity. JCI insight, 6(9), e141486. https://doi.org/10.1172/jci.insight.141486
- Verta, J. P. et al. (2021). Genetic Drift Dominates Genome-Wide Regulatory Evolution Following an Ancient Whole-Genome Duplication in Atlantic Salmon. Genome biology and evolution, 13(5), evab059. https://doi.org/10.1093/gbe/evab059
- Kron, N. S. et al. (2021). Co-expression analysis identifies neuro-inflammation as a driver of sensory neuron aging in Aplysia californica. PloS one, 16(6), e0252647. https://doi.org/10.1371/journal.pone.0252647
- Alvarez-Guaita, A. et al. (2021). Phenotypic characterization of Adig null mice suggests roles for adipogenin in the regulation of fat mass accrual and leptin secretion. Cell reports, 34(10), 108810. https://doi.org/10.1016/j.celrep.2021.108810
- Hall, Z. et al. (2021). Lipid Remodeling in Hepatocyte Proliferation and Hepatocellular Carcinoma. Hepatology (Baltimore, Md.), 73(3), 1028–1044. https://doi.org/10.1002/hep.31391
- Viana, J. et al. (2020). Clozapine-induced transcriptional changes in the zebrafish brain. NPJ schizophrenia, 6(1), 3. https://doi.org/10.1038/s41537-019-0092-x
- Harjula, S. E. et al. (2020). Characterization of immune response against Mycobacterium marinum infection in the main hematopoietic organ of adult zebrafish (Danio rerio). Developmental and comparative immunology, 103, 103523. https://doi.org/10.1016/j.dci.2019.103523
- Ballesteros, I. et al. (2020). Co-option of Neutrophil Fates by Tissue Environments. Cell, 183(5), 1282–1297.e18. https://doi.org/10.1016/j.cell.2020.10.003
- Mehtonen, J. et al. (2020). Single cell characterization of B-lymphoid differentiation and leukemic cell states during chemotherapy in ETV6-RUNX1-positive pediatric leukemia identifies drug-targetable transcription factor activities. Genome medicine, 12(1), 99. https://doi.org/10.1186/s13073-020-00799-2
Fotakis, G. et al. (2020). NeoFuse: predicting fusion neoantigens from RNA sequencing data. Bioinformatics (Oxford, England), 36(7), 2260–2261. https://doi.org/10.1093/bioinformatics/btz879
- Lu, Y. et al. (2020). Interleukin-33 Signaling Controls the Development of Iron-Recycling Macrophages. Immunity, 52(5), 782–793.e5. https://doi.org/10.1016/j.immuni.2020.03.006
- Polinski, J. M. et al. (2020). Unique age-related transcriptional signature in the nervous system of the long-lived red sea urchin Mesocentrotus franciscanus. Scientific reports, 10(1), 9182. https://doi.org/10.1038/s41598-020-66052-3
Binenbaum, I. et al. (2020). Container-aided integrative QTL and RNA-seq analysis of Collaborative Cross mice supports distinct sex-oriented molecular modes of response in obesity. BMC genomics, 21(1), 761. https://doi.org/10.1186/s12864-020-07173-x
- Kron, N. S. et al. (2020). Changes in Metabolism and Proteostasis Drive Aging Phenotype in Aplysia californica Sensory Neurons. Frontiers in aging neuroscience, 12, 573764. https://doi.org/10.3389/fnagi.2020.573764
- Gao, Y. et al. (2020). LKB1 Represses ATOH1 via PDK4 and Energy Metabolism and Regulates Intestinal Stem Cell Fate. Gastroenterology, 158(5), 1389–1401.e10. https://doi.org/10.1053/j.gastro.2019.12.033
- Viiri, L. E. et al. (2019). Extensive reprogramming of the nascent transcriptome during iPSC to hepatocyte differentiation. Scientific reports, 9(1), 3562. https://doi.org/10.1038/s41598-019-39215-0
- Pölönen, P. et al. (2019). Hemap: An Interactive Online Resource for Characterizing Molecular Phenotypes across Hematologic Malignancies. Cancer research, 79(10), 2466–2479. https://doi.org/10.1158/0008-5472.CAN-18-2970
- Adriaenssens, A. E. et al. (2019). Glucose-Dependent Insulinotropic Polypeptide Receptor-Expressing Cells in the Hypothalamus Regulate Food Intake. Cell metabolism, 30(5), 987–996.e6. https://doi.org/10.1016/j.cmet.2019.07.013
- Roberts, G. P. et al. (2019). Comparison of Human and Murine Enteroendocrine Cells by Transcriptomic and Peptidomic Profiling. Diabetes, 68(5), 1062–1072. https://doi.org/10.2337/db18-0883
- Bénéchet, A. P. et al. (2019). Dynamics and genomic landscape of CD8+ T cells undergoing hepatic priming. Nature, 574(7777), 200–205. https://doi.org/10.1038/s41586-019-1620-6
- Moreau, P. R. et al. (2018). Transcriptional Profiling of Hypoxia-Regulated Non-coding RNAs in Human Primary Endothelial Cells. Frontiers in cardiovascular medicine, 5, 159. https://doi.org/10.3389/fcvm.2018.00159
Escobar, G. et al. (2018). Interferon gene therapy reprograms the leukemia microenvironment inducing protective immunity to multiple tumor antigens. Nature communications, 9(1), 2896. https://doi.org/10.1038/s41467-018-05315-0
Norelli, M. et al. (2018). Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nature medicine, 24(6), 739–748. https://doi.org/10.1038/s41591-018-0036-4
Ollila, S. et al. (2018). Stromal Lkb1 deficiency leads to gastrointestinal tumorigenesis involving the IL-11-JAK/STAT3 pathway. The Journal of clinical investigation, 128(1), 402–414. https://doi.org/10.1172/JCI93597
- Bouvy-Liivrand, M. et al. (2017). Analysis of primary microRNA loci from nascent transcriptomes reveals regulatory domains governed by chromatin architecture. Nucleic acids research, 45(17), 9837–9849. https://doi.org/10.1093/nar/gkx680
- Sin, C. et al. (2016). Quantitative assessment of ribosome drop-off in E. coli. Nucleic acids research, 44(6), 2528–2537. https://doi.org/10.1093/nar/gkw137
Contact us
Leave your email address here with a brief description of your needs, and we will contact you to get things moving forward!
