RNA-Seq Expression Analysis: 5 Dirty Secrets
RNA-sequencing-based gene expression analysis has become a staple in biological research, and it is the most common type of analysis we perform at Genevia Technologies. A well-designed RNA-seq experiment provides a rich source of data to address hypotheses and generate new ones. Due to the popularity of RNA seq, countless computational tools and and databases have been developed to enable more and more sophisticated analyses. However, there are important caveats to keep in mind when interpreting the results. We discuss five of them here.
Patterns deceive
Based on the upper panel of the figure below, would you say the two sample groups are clearly distinct? The principal component analysis and clustered heatmap suggest they do. The bottom panel, on the other hand, shows no separation. However, the plots in both panels are based on the same exact simulated expression data, with no systematic difference between the two sample groups. The only difference in the two panels is that the upper one is based on those genes whose expression differs the most between the groups — due to pure noise, in this case —, whereas the lower panel shows the same plots based on all genes (PCA) and the genes which vary the most across all samples regardless of group labels (heatmap).
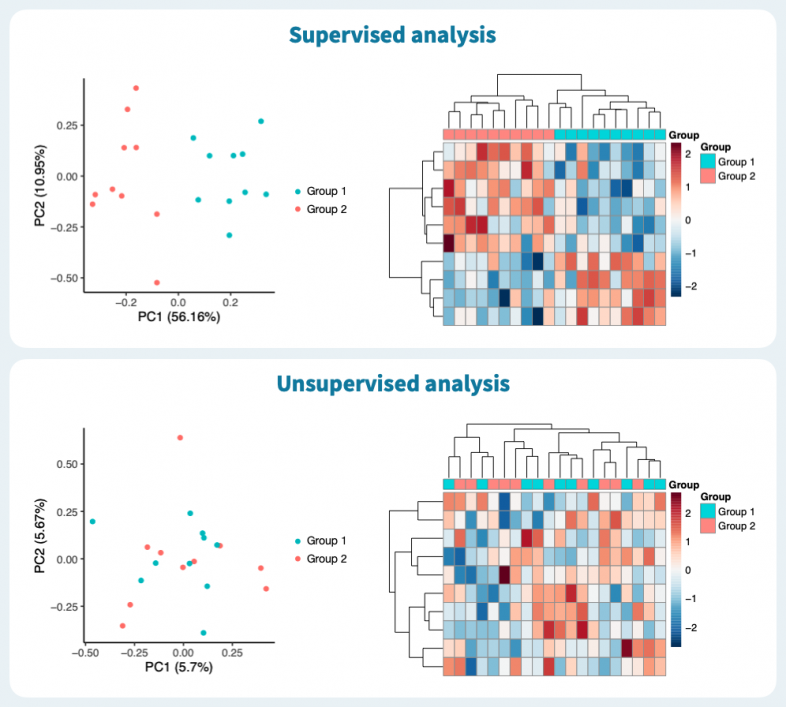
The upper panel represents supervised analysis, in which group labels are incorporated in the workflow, and the lower represents unsupervised analysis, in which group labels are only used to label the data in the plots. To find out whether two sample groups have distinct expression profiles, the unsupervised approach answers our question. Too often, the two approaches are confused, leading to misleading visualizations and wrong conclusions. That said, a supervised analysis is great for identifying biomarkers, for instance, but one must ensure the analysis answers the right question.
All genes are significant
When comparing two conditions with any true biological difference, such as healthy vs normal samples or treated vs untreated samples, any given gene is likely to differ expression-wise, if only slightly. However small the difference, it will become statistically significant as the sample size increases. Especially with large sample sizes, it is important to also consider the effect size, or expression fold change. Even so, any cutoff for both the statistical significance and fold change in determining differentially expressed (DE) genes is more or less arbitrary.
A binary grouping of genes into DE or non-DE may help in interpreting the data, but it is important to acknowledge that it is a simplification. For downstream analysis, it is often possible to avoid this information-reducing simplification and retain higher data fidelity. Gene set enrichments, for instance, can be obtained based on a ranking of all genes by their test statistic or fold change rather than by an over-representation analysis of a hard set of DE genes, obviating the need to specify arbitrary cutoffs.
Not all positives are true
Continuing on the previous point, it is important to note that not only does a binary grouping of genes into DE or non-DE result in some meaningful genes being filtered out (false negatives), but it also results in false positives. Assuming we have defined a threshold for both statistical significance and expression fold change, any gene that makes the cut may do so simply because of noise rather than signal.
The good news is that the number of such false positive genes can be controlled. In a DE analysis, p-values are adjusted based on their distribution. Commonly, this involves specifying a desired false discovery rate (FDR; the fraction of false positives among all DE genes). For instance, with an FDR cutoff of 5% we may expect that one in twenty DE genes is “false” DE, but we do not know just which one it is.
To summarize, a list of differentially expressed genes is not an exclusive and complete list of “true” genes relevant to the underlying comparison, but rather a shortlist of genes which are likely, but not guaranteed, to be the most relevant ones.
RNA is not protein
What is the functional gene product? For most genes, it’s a protein, not RNA. With RNA-sequencing data, it is good to keep in mind that any quantified difference is that of RNA levels rather than functional proteins.
Depending on the organism, tissue and gene, mRNA concentrations may correlate poorly with protein concentrations. Even when the correlation is high, the activity of the protein may be modulated by post-translational modifications such as phosphorylation. Changes in protein concentrations and activities may go unnoticed in RNA-seq data, unless they affect the mRNA levels of other genes, as transcription factors do. While clearly distinct cellular states are bound to show up in RNA-seq, potential RNA-protein discrepancies must be acknowledged especially when the end goal is to identify individual proteins to use as drug targets or biomarkers, for instance.
Pathway analysis is just as good as the used pathway database
Results from a DE analysis are typically interpreted with the aid of pathway analyses. Simpler analyses involve annotating a gene list with molecular processes or phenotypes which are known associate with the DE genes, using Gene Ontology (GO) terms, for instance. More in-depth pathway analyses integrate the expression data with the network of known interactions between genes or gene products, such as Ingenuity Pathway Analysis (IPA). This results in a more mechanistic interpretation of the differences between sample groups, rather than just a list of GO terms.
Any gene set or pathway analysis, however, is imperfect. Just like in the DE analysis, any list of pathways will contain false positives and miss some pathways as false negatives. The reasons for this include previously discussed noise and transcriptome-proteome discrepancies, but also limitations and biases in the used pathway databases.
The data that has been used to define pathways are more extensive in model organisms — especially mouse and human — meaning that the idiosyncrasies of other organisms may be poorly represented. Furthermore, biological systems that are easier to sample and study, such as cell lines rather than tissue biopsies, are likely to be over-represented, as well as ones that are studied more for clinical and funding reasons, such as cancer. Pathway analyses with non-cancer samples often result in cancer-related pathways simply because the genes in question have been discovered and annotated in the context of cancer. Especially in rare conditions with previously uncharacterized pathway behavior, the power of a priori annotated pathways is limited.
Summary
As a bioinformatics service provider, we believe it is not our responsibility to just analyze the data, but also to help our customers interpret the results. Here we discussed some general points, but each data set and analysis comes with its own strengths and limitations. Whoever analyzes your RNA-seq data, just make sure they have the experience and time to discuss the outcome of the analysis and help you in drawing the right conclusions. Just being aware of the caveats may save a lot of time!
Learn more
Short case studies of projects in which we analyzed RNA-seq data to help our customers to evaluate EV isolation protocols, discover biomarkers for macrophage-based cell therapy and study the mechanism of action of a drug.
Papers in which we helped our customers with various types of expression data analysis on topics such as extracellular nanovesicles, hepatocellular carcinoma, prostate cancer, alzheimer’s disease, schizophrenia, psychopathy, endometriosis, osteoblast differentiation and crop protection. See also papers which our team members have co-authored.
Read more about our RNA-seq analysis service.