Computational Neoantigen Prediction: Unlocking Personalized Immunotherapy
The concept of neoantigens—mutated peptides uniquely presented by tumor cells—has redefined the frontier of cancer immunotherapy. These tumor-specific epitopes hold the promise of highly personalized treatment approaches, ranging from therapeutic vaccines to adoptive T-cell therapies. But how does one get from DNA and RNA to identifying neoantigens? This is where computational neoantigen prediction comes into play—a rapidly evolving field at the intersection of genomics, transcriptomics, immunology, and artificial intelligence.

The Basics: What Are Neoantigens?
Neoantigens are novel peptide sequences that arise specifically in cancer cells. These peptides are processed intracellularly and presented on HLA class I molecules — and, in antigen-presenting cells and some tumors, in HLA II as well. Recognized by T cells, neoantigens can elicit potent anti-tumor immune responses.
Cancer cells express different types of antigens:
- Tumor-specific antigens — synonymous with neoantigens — are unique to cancer cells and absent in healthy tissues, making them especially attractive for immunotherapy.
- Tumor-associated antigens and cancer germline antigens, which are also found in some normal tissues, raising concerns about immune tolerance and off-target toxicity.
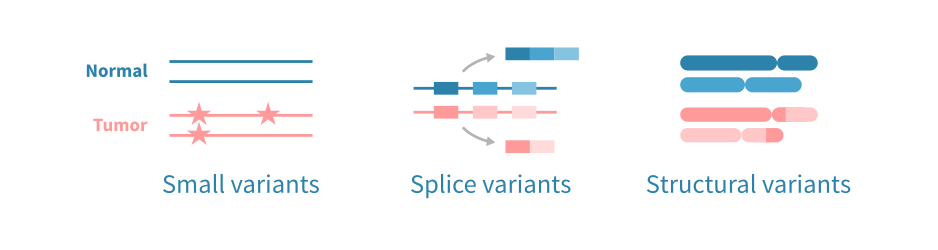
Neoantigens primarily originate from non-synonymous somatic mutations, including single nucleotide variants (SNVs), small insertions/deletions (indels), and gene fusions. They may also arise from post-transcriptional events, such as alternative splicing, intron retention, or premature transcription termination. In addition, cancer-specific post-translational modifications (e.g., phosphorylation, glycosylation) represent a lesser-studied but potentially important source of neoantigens.
The Pipeline: From Mutation to Neoantigen
For most neoantigen discovery needs, a modular and customizable pipeline is often the most effective approach. This flexibility is essential, as the optimal methods and data types vary depending on the class of neoantigen being studied.
The workflow for computational neoantigen prediction can be divided into four main steps:
1. Identification of Neoantigenic Mutations
This step is critical and often the most challenging. The question isn't just “Which tool should I use?” but rather “What kind of neoantigens am I trying to uncover?” Neoantigens arise from different types of mutations:
- SNVs and small indels. These small mutations are detected using high-quality whole-exome (WES) or whole-genome sequencing (WGS) data, with variant callers such as Mutect2 or Strelka2.
- Alternative splicing (AS) variants. AS-derived neoantigens arise from transcriptome-level events like exon skipping and intron retention, making tumor RNA-sequencing essential for their detection. These neoantigens are particularly relevant in tumors with low mutational burden but altered regulation of splicing. Approaches to detect splice variants include:
- Isoform-based tools (e.g., Cufflinks, DiffSplice)
- Read count-based tools (e.g., SplAdder, SUPPA2)
- Gene fusions. Gene fusions arise from chromosomal rearrangements, such as translocations, which can produce immunogenic chimeric proteins. These structural variants are best detected using RNA-seq or WGS data, with RNA-seq -based detection carrying the additional benefit of ensuring that the fusion gene is transcribed. Approaches for fusion detection include:
- Assembly-first methods (e.g., TrinityFusion)
- Mapping-first methods (e.g., Arriba, STAR-Fusion)
2. HLA Typing
HLA molecules present peptides to T cells. HLA genes are highly polymorphic, and each gene and specific allele thereof will present certain peptides better than others. Thus, accurate HLA typing is essential in predicting whether a neoantigen is likely to be presented or not. Both class I and class II molecules are relevant, with class II gaining increasing attention.
HLA typing can make use of WES, WGS or RNA-seq data, and is based on aligning sequencing reads to know HLA alleles from a database such as IPD-IMGT/HLA, followed by scoring or optimization methods to infer the best-matching alleles. Tools such as OptiType and HLA-HD can be used. HLA typing can be challenging due to high polymorphism, incomplete allele sequences and difficulties in detecting rare variants. Long-read sequencing or targeted HLA sequencing can help address these limitations.
3. Peptide Presentation: HLA Binding Prediction
This step assesses whether a mutant peptide will bind to HLA molecules—an essential prerequisite for T-cell recognition. Tools that model peptide-HLA binding, such as MHCflurry or NetMHCpan are used. These tools typically use machine learning models (especially artificial neural networks) that are trained on binding affinity and immunopeptidomics data to capture the complex, non-linear interactions between peptides and HLA molecules.
HLA allele-specific predictors perform well for common alleles, while pan-allele models extend predictions to rare or uncharacterized variants. Despite the progress, challenges remain due to limited training data on rare alleles and the complexity of immunopeptidomics. Hybrid methods and monoallelic strategies are used to improve prediction accuracy.
4. Filtering and Prioritization of Neoantigens
Early neoantigen filters focused on HLA binding affinity (e.g., IC50 < 500 nM), but this alone is not predictive of immunogenicity. Modern strategies integrate multiple features, such as clonality, amino acid properties, peptide entropy, and HLA promiscuity into machine learning models to predict immunogenicity.
These predictive models face challenges such as sparsity in training data and overfitting. The most recent frameworks distinguish between presentation (e.g., HLA binding affinity, expression, stability) and recognition (e.g., agretopicity, foreignness) features for more accurate neoantigen prioritization.
Optional Enhancements
- RNA-seq validation – If mutations are identified from DNA-sequencing data, RNA-sequencing can be used to confirm that the predicted neoantigenic mutations are expressed, at least on the trasncirpt level.
- Tumor clonality analysis – Tools like PyClone or SciClone distinguish clonal from subclonal mutations.
- Immunogenicity ranking – Tools like PRIME and DeepHLApan predict T-cell recognition probability, not just the HLA binding affinity.
- TCR binding prediction – Emerging tools include ERGO, TCRmatch, and NetTCR.
The Pitfalls: Key Considerations And Challenges
- False positives are common: Binding does not equal immunogenicity
- Tumor heterogeneity: Neoantigens may be subclonal and not universally expressed – limiting any clinical benefit from a neoantigen-based therapy
- HLA diversity: Limited training data reduces the power to predict neoantigens for patients with rare HLA alleles
- Validation is critical: In silico predictions must be confirmed experimentally
A Data-Driven Era for Personalized Immunotherapy
Computational neoantigen prediction is revolutionizing the design of personalized cancer immunotherapies. While experimental validation remains indispensable, neoantigen prediction is increasingly robust, modular, and AI-powered—paving the way toward individualized therapies.
Although current tools (e.g., nextNEOpi, ScanNeo2) are primarily built for bulk sequencing data, their modular architecture allows adaptation to single-cell datasets (scRNA-seq, scDNA-seq). In this rapidly evolving landscape, skilled bioinformaticians are essential—not only to navigate complex data, but also to develop and customize computational pipelines tailored to specific research needs.
The author is a co-developer of the above-mentioned nextNEOpi. Learn more about the pipeline on github or the article.
Contact us
Leave your email address here with a brief description of your needs, and we will contact you to get things moving forward!
